

日本研究人员开发出一种新型计算框架,能够从高速原子力显微镜(HS-AFM)的地形成像数据中推断动态蛋白质的三维原子模型。该技术由金泽大学纳米生命科学研究所的Holger Flechsig与名古屋大学的Florence Tama共同领导的研究团队提出,相关成果已发表于《ACS Nano》期刊。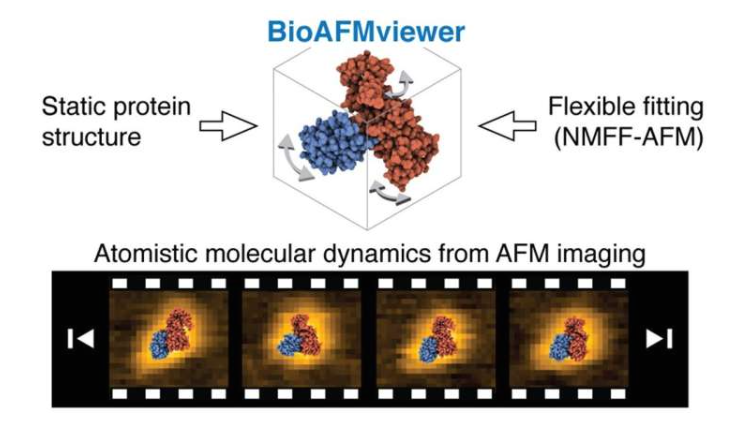
高速原子力显微镜是当前能够直接观察蛋白质动态活动的实验技术。然而,其空间分辨率存在局限,难以直接提供原子层面的生物分子功能信息。为突破此限制,研究团队采用了一种计算效率较高的灵活拟合方法,该方法由Tama团队开发,能够模拟已知静态蛋白质结构的构象动力学,从而识别最匹配实验原子力显微镜图像的原子模型。
该计算框架已集成至由Flechsig团队维护的BioAFMviewer软件平台中,为分析原子力显微镜成像数据提供了直接的工作流程。研究团队对多种蛋白质的高速原子力显微镜数据进行分析,结果显示灵活拟合方法能够推断包含大振幅运动的原子模型,显著提升了对分辨率受限测量中功能构象动力学的理解。
BioAFMviewer软件具有较高的计算效率,可应用于大型蛋白质组装体。研究团队以一条由约28万个原子组成的4兆道尔顿肌动蛋白丝为例,展示了该方法的有效性。基于高速原子力显微镜拓扑电影数据,团队成功重建了涉及功能性构象转变的蛋白质动力学原子分子电影。
该软件实现了计算效率较高的灵活拟合,将现有结构数据和分子建模与实验相结合,为单分子成像数据的大规模分析提供了新的可能性。通过充分利用高速原子力显微镜的解释能力,该技术有望促进对纳米级生物过程的深入理解。
正则模式柔性拟合原子力显微镜方法采用计算效率较高的迭代正则模式分析,模拟大振幅构象变化,从而识别最能代表测量形貌图像的动态原子模型。BioAFMviewer项目由Holger Flechsig于2020年发起,旨在集成高分辨率生物分子结构和建模数据,用于分析分辨率受限的原子力显微镜测量。该软件提供用户友好的交互界面,支持生物分子可视化及模拟原子力显微镜成像,其灵活拟合方法性能通过并行计算得到显著提升。
更多信息: Romain Amyot 等人,通过高速原子力显微镜成像,灵活拟合推断生物分子大振幅构象动力学的原子精度模型,ACS Nano (2025)。期刊信息: ACS Nano