

西湖大学生命科学学院杨剑教授带领的研究团队开发出一种泛基因组引导的基因组组装方法。该方法结合经济高效的长读长和短读长混合测序策略,成功构建了超过1000个个体的泛基因组,克服了以往小样本泛基因组的局限,为医学和群体遗传学研究提供了关键基础设施。相关成果已发表在《自然》杂志上。
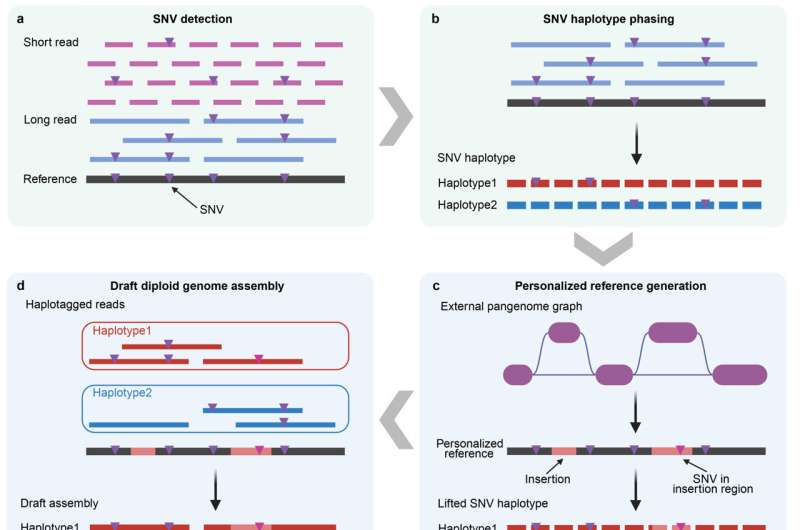
人类基因组计划完成后,单一线性参考基因组成为生物医学研究的基础,但无法全面反映人群间的遗传多样性,导致传统分析常忽略结构变异和串联重复等复杂遗传变异。为此,研究人员提出泛基因组概念,即代表人群遗传多样性的基因组序列集合。尽管长读长测序技术进步,但高昂成本使先前泛基因组样本量仅几十个个体,难以准确估计变异频率或解析低频变异和高复杂度区域,开发适用于大规模群体的经济高效泛基因组构建策略成为迫切需求。
杨剑团队长期致力于统计遗传学、基因组学和人类复杂性状大数据分析的方法学研究,通过开发高效计算方法攻克大规模基因组数据处理挑战,其分析工具如GCTA-GREML、SMR和gsMap已在全球广泛应用。为应对构建大规模泛基因组的挑战,团队开发了泛基因组引导的基因组组装工作流程,采用泛基因组引导框架整合整个队列序列信息,利用基于适度覆盖度的Illumina短读长和PacBio长读长全基因组测序数据的混合测序策略,显著降低测序成本,同时从适度覆盖度数据中组装基因组,为未来群体规模的混合测序研究提供实用新技术途径。
应用该方法,研究团队构建了目前全球最大的人类泛基因组,包含1116个平均质量值为46的二倍体基因组,识别出4.053亿个碱基对的非参考序列,其中2620万个碱基对序列被注释为功能性基因和预测调控元件,扩展了对人类基因组非参考序列的理解。利用大规模组装数据集,研究人员编制了全面的遗传变异目录,包括3540万个小型变异以及110,530个结构变异、485,575个串联重复和86万个嵌套变异。使用该目录,团队在多个尺度上表征了医学相关变异,如改变基因的结构变异和致病性串联重复扩展,表明该目录为致病突变临床筛查提供重要参考。通过整合基因表达数据,团队识别出3256个涉及复杂变异的表达数量性状位点,阐明了这些变异的调控复杂性。该研究推进了对复杂遗传变异及其功能影响的理解,为人类健康研究和其他物种泛基因组研究建立了新范式。
出版详情:作者:Westlake University;标题:《Assembling more than 1,000 human genomes affordably: New method could power genetic screening's future》;发表于:《Nature》(2026);杂志信息: Nature