

维度网讯,美国橡树岭国家实验室(ORNL)与IBM Quantum、克利夫兰诊所等机构联合发表论文,首次将量子计算运用于聚变包层关键材料FLiBe中氚化学行为的电子结构计算,展示了量子-经典混合计算框架对该类复杂体系进行高精度模拟的能力。
当前主流磁约束聚变路线主要依赖氘-氚(D-T)反应。论文指出,一座1GW级聚变电站每天大约需要消耗0.5 kg氚,而全球现有氚库存约25 kg。未来商业聚变需要依靠包层中的锂元素通过中子反应实现氚增殖,并高效回收重新注入等离子体。因此,理解氚在包层材料中的生成、迁移和结合行为,是聚变工程绕不开的核心问题。
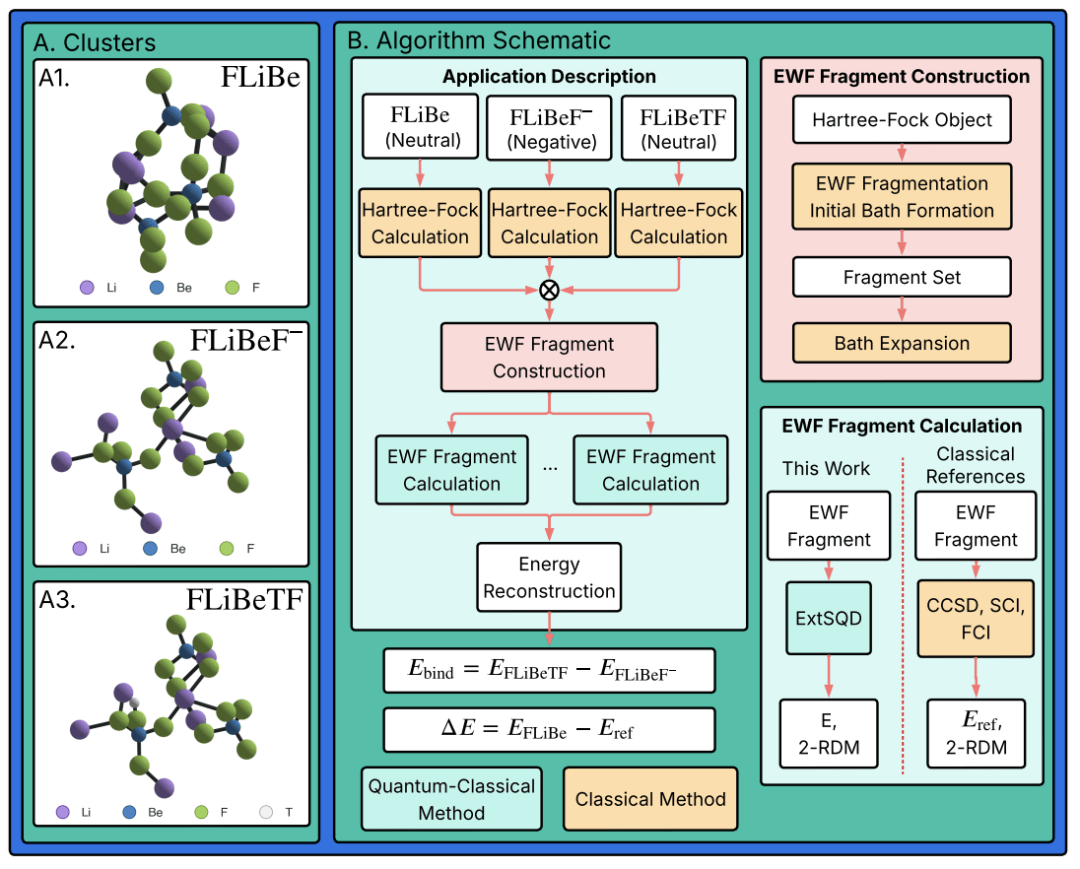
FLiBe(LiF-BeF₂)熔盐被认为是先进聚变堆的重要候选材料,它含锂可通过中子反应产氚,且液态运行可同时承担传热与燃料增殖功能。但氚在高温熔盐环境中可能以氚离子(T⁺)、氚分子(T₂)或与氟形成复杂结构(F-T-F)等不同形态存在,这些形态直接关系到氚的停留时间、提取效率和循环成本。
解氚在FLiBe中的行为在计算上是一个难题。传统方法如密度泛函理论(DFT)、分子动力学(MD)模拟和机器学习势函数(MLFF)在处理含氚的复杂熔盐分子及带电离子团簇这类强电子相关体系时存在精度瓶颈。更高精度的完全组态相互作用(FCI)或耦合簇方法,其计算成本又会随体系规模呈指数级激增,难以用于真实聚变材料环境。
此次研究发表在arXiv预印本平台,论文题为《聚变包层熔盐的量子计算》(Quantum Computations on Fusion Blanket Molten Salts)。研究人员聚焦于氚与包层材料的结合机制,利用量子-经典嵌入波函数计算框架,对9种不同FLiBe分子构型进行计算。他们并未直接模拟宏观包层,而是通过“扩展基于样本的量子对角化”(ext-SQD)算法,将复杂片段交由IBM Heron量子处理器求解,最终与经典高精度FCI结果对比。
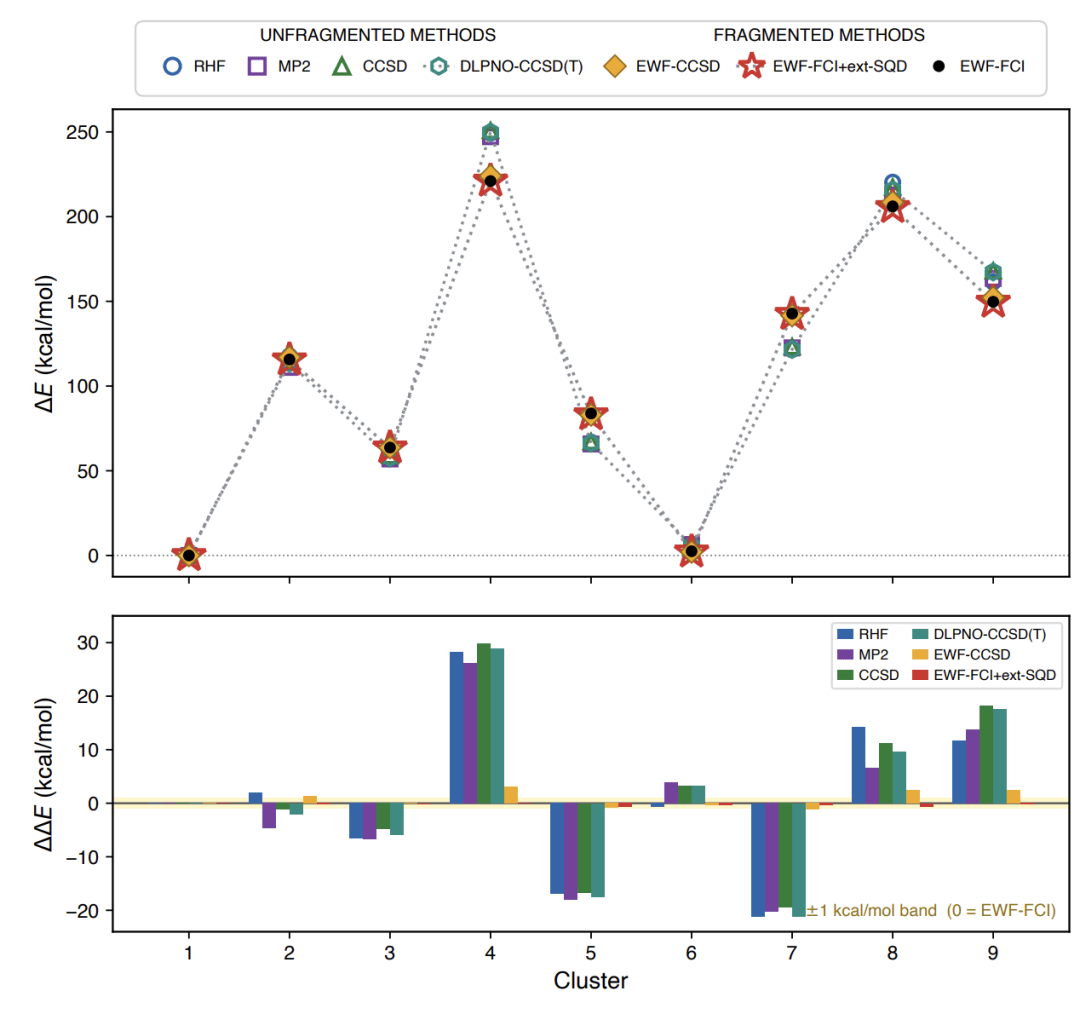
结果显示,ext-SQD量子方法与FCI结果的偏差约0.7 kcal/mol,平均绝对偏差约0.3 kcal/mol。这是业界首次在带电离子体系,特别是无机熔盐体系中,成功实现量子-经典计算验证。
这项研究标志着聚变材料研发有望从依赖实验试错的传统模式,迈入基于高性能计算、AI模型与量子计算的材料基因组计算设计阶段。过去,包层材料开发依赖于漫长的“实验试错—中子辐照测试—宏观性能评价”周期。未来,借助高性能计算、AI与量子计算的融合,研究人员可能从原子甚至量子层面精准预测熔盐组合对氚释放的适配性。同时,该研究也揭示了氚燃料循环正从宏观的系统工程问题下沉为底层的材料科学问题。
该论文为量子计算用于聚变材料研究打开了技术路径。同时,研究也指出,现有量子计算规模尚不足以涵盖宏观尺度的工程情况,未来需要更大规模量子计算体系和底层算法的进一步突破。此次ORNL、IBM Quantum和克利夫兰诊所的跨界合作,提供了一条值得关注的技术融合方向。










