
维度网讯,日本东北大学(Tohoku University)的研究人员发现一种新的催化剂设计原理,揭示了双原子催化剂(dual-atom catalysts, DACs)在氧还原反应中呈现“双萨巴捷最优”(dual-Sabatier optima)模式,挑战了沿用数十年的单峰火山模型假设,有望为降低氢燃料电池成本提供新路径。
燃料电池被视作构建低碳社会的关键装置,其通过氢气发电且排放清洁。但许多燃料电池仍依赖铂等贵金属来驱动氧还原反应(ORR),这一过程直接影响性能和成本。传统催化理论以“单峰火山”模型解释活性规律,认为最佳催化剂位于化学性质的狭窄区间内。然而,研究团队在分析来自数字催化平台(Digital Catalysis Platform, DigCat)的大规模实验数据集时发现,双原子催化剂并未遵循这一预期模式。
研究人员运用先进理论模拟、微动力学建模(microkinetic modeling)和机器学习,对超过200种双原子催化剂展开研究。结果显示,DACs主要受一种称为解离机制(dissociative mechanism)的反应路径控制,而非单原子催化剂中常见的缔合机制(associative mechanism)。这一变化对催化剂活性表现产生重大影响:DACs不再呈现单一最佳性能峰,而是显示出两个分离的最优区域,即“双萨巴捷最优”。两个峰的出现源于反应过程中限速步骤的转移,在氧解离、氧质子化和羟基质子化之间切换。
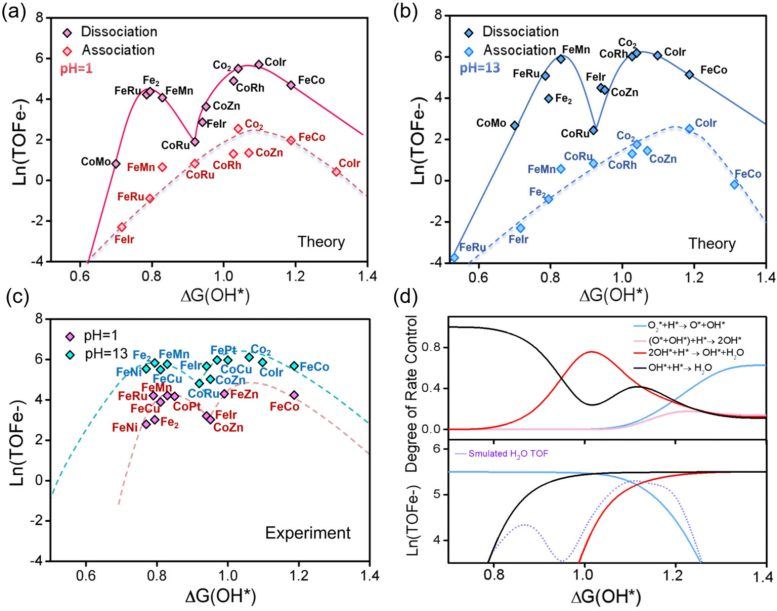
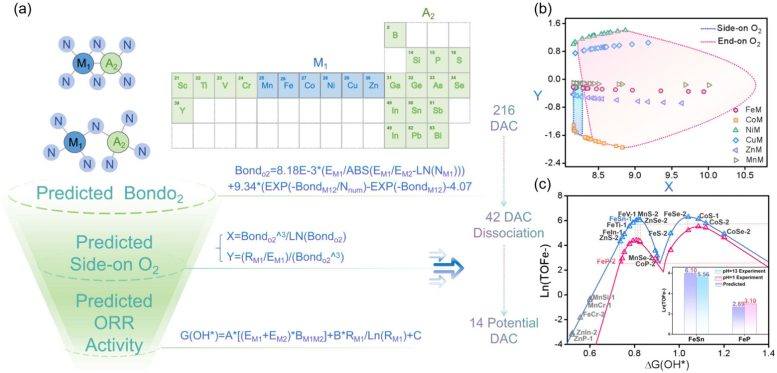
研究人员发现,这一原理适用于多种催化剂类型,涵盖由过渡金属、类金属元素甚至非金属原子构成的体系。通过结合可解释机器学习(interpretable machine learning)与理论建模,团队构建了一个预测框架,能够快速识别有前景的催化剂结构。东北大学先进材料研究所(WPI-AIMR)杰出教授Hao Li表示,长期以来的假设认为双原子催化剂与单原子催化剂遵循相同活性规则,而最新工作表明,当两个原子协同合作时,可能涌现出完全不同的机制,为设计清洁能源技术用的高效材料开辟了新机遇。
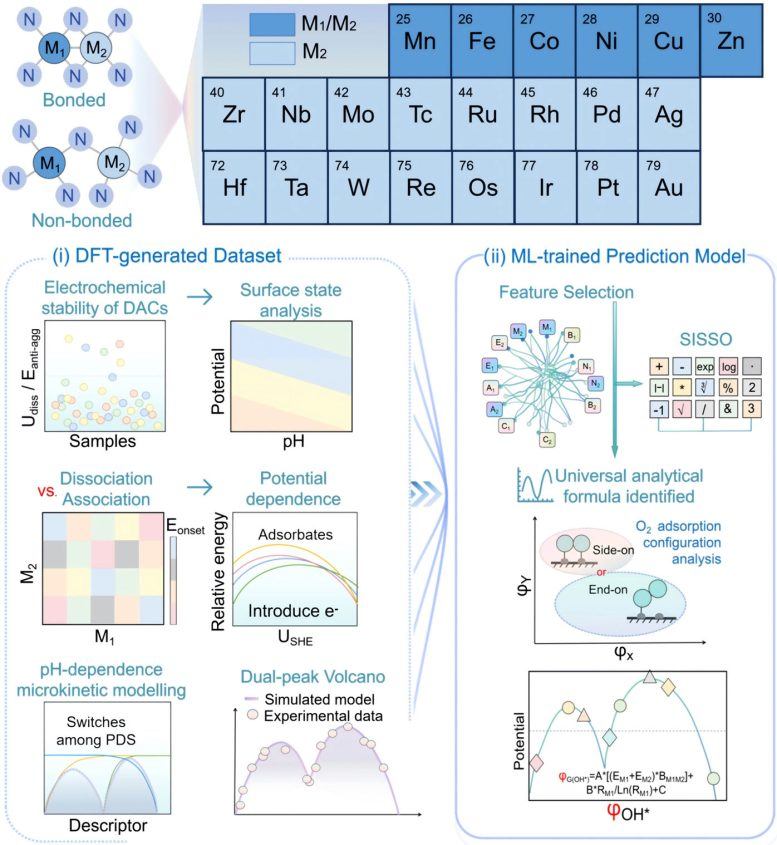
该发现的潜在影响可能超出燃料电池范畴,或能为其他能量转换和化学生产过程的催化剂开发提供指导。研究还展示了人工智能如何从现有实验数据中提取隐藏的科学规律,从而加速新材料筛选。接下来,团队计划将该方法应用于更复杂的多金属催化剂,以及ORR之外的其他能源相关反应,并通过将AI代理、机器学习和电化学模拟整合进DigCat平台,创建一个完全自主的数字系统,用于面向可持续能源的下一代催化剂快速设计。









