
MHRA发布临床试验体外诊断提交新规,2026年4月起实施
2026-02-05 10:23
收藏
英国药品和医疗产品监管局(MHRA)近日宣布,将规范临床试验中体外诊断(IVD)和伴随诊断(CDx)的审查流程。根据新规,自2026年4月28日起,申办者必须在研究性药品临床试验(CTIMP)提交材料中提供所用体外诊断设备的详细信息,取代此前零散的指南要求。若不遵守新规,相关试验将无法继续进行。

目前,英国的临床试验需经过MHRA和伦理委员会的并行评估。MHRA临床试验负责人Kingyin Lee在网络研讨会上表示:“我们计划将体外诊断设备评估纳入这一联合审查流程。这是20年来临床试验法规最重要的更新,旨在提升患者安全、增强试验透明度,并构建一个简化、灵活且相称的监管框架。”
根据新要求,若体外诊断设备已获得UKCA或CE认证,并在试验中按市场准入要求使用,申办者需通过综合研究申请系统(IRAS)在CTIMP提交中说明相关细节。若未获得认证,则需提交健康研究所豁免(HIE)。此外,在北爱尔兰使用的体外诊断设备必须注明其欧盟医疗器械法规(EU MDR)指定。
除上述信息外,申办者还需通过IRAS提交分析性能研究报告或表格摘要。MHRA低风险评估经理Anthony Carter指出:“作为体外诊断审查流程的一部分,我们将设置最长七天的验证窗口。未提供所需体外诊断文件的申请将被视为无效,以确保联合审查流程顺利进行。”Carter还强调,申办者必须在CTIMP提交中包含封面信,明确说明试验中使用了体外诊断设备,以保障系统流程的规范性。
MHRA表示,通过将体外诊断评估与临床试验审查整合并提供单一结果,新框架有望减少重复工作,提升使用体外诊断的临床试验的透明度。体外诊断在临床试验中的作用日益凸显,可用于样本检测、患者分层、治疗反应监测等多个环节,为临床决策提供关键数据支持。
相关推荐

新加坡Gero获1700万美元融资,加速衰老药物研发
2026-06-20

Clarify Health收购Loyal Health,整合超8万提供者档案与患者激活平台
2026-06-20

美国宾夕法尼亚大学AI从朊病毒中发现抗生素候选
2026-06-20

法国Ethylowheel融资100万欧元,皮肤测酒设备销60国
2026-06-19
美国Memento完成9300万美元A轮融资,获双抗全球许可
2026-06-19
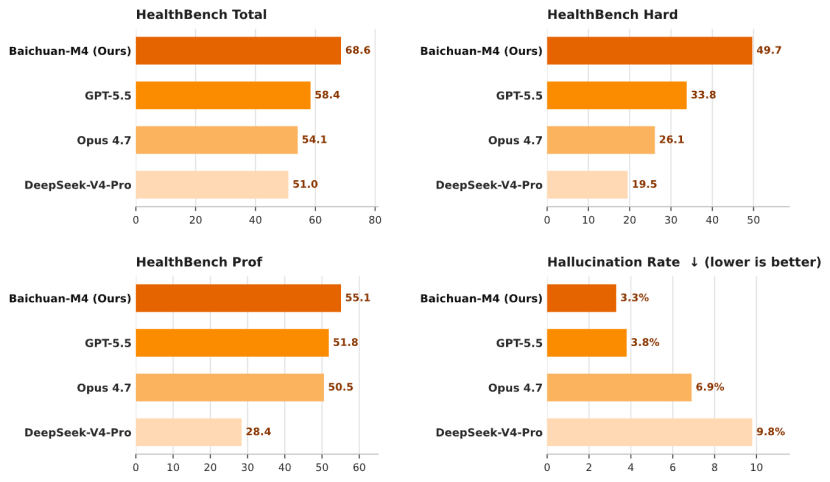
中国百川智能M4医疗大模型发布,综合得分68.6
2026-06-19

美国医疗技术公司Channel Robotics融资460万美元
2026-06-19

美荷团队获900万美元资助研发CAA分子影像技术
2026-06-19
澳大利亚Memphasys签署Monash IVF全国独家供应协议
2026-06-18

美国医疗科技公司Icarus Medical获720万美元A轮融资
2026-06-18