
Argenica公司卒中药物获EMA儿科豁免,简化欧洲审批路径
2026-02-03 17:31
收藏
Argenica Therapeutics在欧洲监管方面取得重要进展,其治疗急性缺血性卒中的候选药物ARG-007获得欧洲药品管理局(EMA)的全面儿科豁免。这一决定为ARG-007在欧洲的审批路径扫除了关键障碍。

根据欧洲法规,制药公司通常需要在提交成人适应症上市授权申请前制定儿科研究计划,但EMA此次授予了产品特定豁免。豁免覆盖所有儿科年龄段,从新生儿到18岁以下人群,这意味着Argenica无需在欧洲进行儿科临床试验即可推进ARG-007的审批。
EMA评估认为,急性缺血性卒中在儿童中罕见,儿科研究既缺乏可行性也缺少科学依据。强制进行儿科试验会增加成本、延误进程并带来复杂性,却无法带来相应的临床益处。这一监管决定显著简化了ARG-007的开发路径,使公司能够专注于成人卒中患者的审批推进。
从开发和商业角度,获得儿科豁免降低了Argenica欧洲战略的风险。原本可能需要复杂全球研究设计、漫长入组时间表和高昂运营成本的儿科卒中试验得以避免,为公司节省了资金和资源。这些资源可转而用于支持成人急性缺血性卒中批准的研究,有助于保持预期的监管时间表。
Argenica董事总经理Liz Dallimore博士表示:“这一决定是我们全球监管战略的关键里程碑,它为ARG-007的开发进展提供了清晰度和确定性。”她补充说,简化后的路径也有望吸引潜在制药合作伙伴的关注,这些合作伙伴正在评估该项目的风险概况和上市时间。
ARG-007是一种神经保护疗法,旨在减少缺血性卒中后的脑组织死亡并限制继发性损伤。获得EMA儿科豁免后,该药物在欧洲的监管路径更加明确,为后续开发奠定了基础。
相关推荐

新加坡Gero获1700万美元融资,加速衰老药物研发
2026-06-20

Clarify Health收购Loyal Health,整合超8万提供者档案与患者激活平台
2026-06-20

美国宾夕法尼亚大学AI从朊病毒中发现抗生素候选
2026-06-20

法国Ethylowheel融资100万欧元,皮肤测酒设备销60国
2026-06-19
美国Memento完成9300万美元A轮融资,获双抗全球许可
2026-06-19
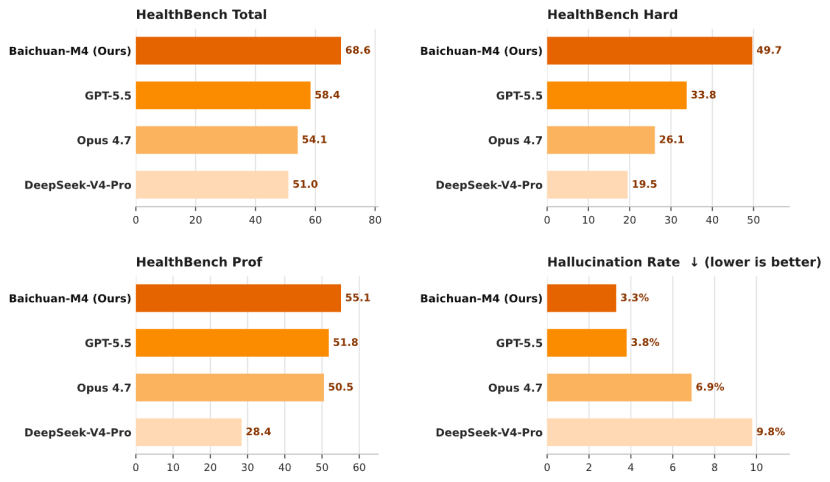
中国百川智能M4医疗大模型发布,综合得分68.6
2026-06-19

美国医疗技术公司Channel Robotics融资460万美元
2026-06-19

美荷团队获900万美元资助研发CAA分子影像技术
2026-06-19
澳大利亚Memphasys签署Monash IVF全国独家供应协议
2026-06-18

美国医疗科技公司Icarus Medical获720万美元A轮融资
2026-06-18